Abstract
The DNA in our cells contains our "blueprints," but it's the proteins in our cells that do most of the work. The Human Genome Project has allowed us to start reading the blueprints, but we still don't understand what most of the proteins do. This is a fairly advanced project that explores ways of identifying the function of unknown proteins.Summary
Edits: Svenja Lohner, PhD, Science Buddies
Sponsor: Molecular Sciences Institute (MSI), Berkeley, California
Management: The Kenneth Lafferty Hess Family Charitable Foundation
Objective
The goal of this project is to learn how to uncover the functions of an unknown protein using computational methods. In doing so, you will learn about the connection between how a protein performs its job(s) and how its corresponding DNA sequence changes throughout evolution. You will also get a chance to explore computational methods for figuring out the specific functions of a protein, based only on its amino acid (protein) sequence. You will choose several proteins to study, and for each of these proteins, you will generate a hypothesis about which parts of the protein are likely to be most important to its function. This will be done by comparing the human version of the protein to other (non-human) versions of the protein. You will then use databases that identify signatures of common functional components of proteins and assess whether or not these analyses are consistent with your hypothesis.Introduction
Proteins are the "building blocks of life"—not only are many of the physical structures of cells made of proteins, but many of the tasks that are essential to life are carried out by proteins. Human beings have genes encoding about 30,000 proteins, yet only a small fraction of these proteins have been studied intensely enough to be well understood. The functions of the vast majority of proteins—found in humans and elsewhere—remain entirely unknown. The first step in figuring out the function of a protein is determining its amino acid sequence. The human genome project (and other genome projects) has achieved this goal for a large number of proteins. Unfortunately, the amino acid sequence alone tells you very little about the function of a protein, and the next steps in figuring out a protein's function are much less clearly defined, and very challenging. But, because scientists stand to gain so much from understanding the functions of a larger number of proteins, a major focus of this generation's scientists has been, and continues to be, figuring out how to learn as much as possible about a protein from its amino acid sequence and its corresponding DNA sequence.One of the first steps that scientists typically take in characterizing a newly discovered protein, is to identify the parts of the protein that are essential to its function. This way, even if they aren't able to figure out what each of those parts do, they can at least focus their future studies on the most important parts of the protein. One way to identify important parts of a protein is to track which parts have remained relatively unchanged throughout evolution. If a random DNA mutation changes the amino acid that it codes for, the likelihood that that particular DNA change will be passed on to future generations depends on whether or not the changed amino acid destroys the function of the protein. If the change occurred in an important part of the protein, and destroyed that protein's function, then that mutated protein is not likely to be passed on throughout evolution. Similarly, if the change occurred in an unimportant part of the protein, and did not affect its function, then that changed sequence is much more likely to be passed on to future generations. Thus, when you compare DNA or protein sequences between two distantly related species, you would expect fewer differences in the "important" parts of the protein and an accumulation of more changes in less important parts. The term "conserved" is used to describe the DNA/RNA/amino acid sequences that remain relatively unchanged throughout evolution, and it is generally assumed that such conservation is an indication of functional importance.
Scientists have learned a lot about the nature of proteins by comparing the same protein across many different species. One of the key findings has been that proteins are often modular—that is, their major job is often accomplished by the combined action of several small functional units. These smaller functional units are called domains. Another key finding is that nature often recycles the same protein domain repeatedly in order to accomplish the same task in very different proteins. Because of this, it has been possible to build databases of sequences (signatures) associated with particular functions. These databases can in turn be used to help figure out the function of an unknown protein based only on its DNA, mRNA or amino acid (protein) sequence. By identifying the domains that a protein contains, it is often possible to figure out what the overall job of the protein is. For example, if a protein contains a DNA binding domain and an estrogen-response domain, it is likely that this protein's job is to control the levels of production of another protein in response to estrogen levels.
In this project you will use one set of tools to identify regions of a protein that are likely to be functionally important, and another set of tools to explore what those functions might be.
Keys:- Spend some time thinking about which proteins to look at, as well as which species to compare. It would be valuable to explore using different "types" of proteins and thus different "parts" of the evolutionary tree for this study. That is, some gene/protein types are found in almost all living eukaryotes (what does this mean?), such as a protein involved in respiration or glycolysis. Therefore, you could get the protein sequence for a glycolysis gene from animals across the animal kingdom from yeast to humans. On the other hand, a protein involved in red blood cell function (like hemoglobin) will probably only be found in animals with red blood cells—therefore a comparison across the whole animal kingdom would be unsuccessful. Both types of studies are equally valuable—and maybe you could choose a couple of each type of protein.
- Spend some time thinking about which species you choose to compare. Much of your species choice will be dictated by how "fundamental" the gene is as well as how many species have published sequences for that gene (very few species have all genes sequenced). But, if you choose a gene that exists in all living organisms, you will have a wide selection of species to choose from. In this case, you should consider choosing a variety of species from different parts of the tree of life. Think about which species are best for each of your proteins of interest.
- You can use mRNA or protein sequences to ask similar questions. How different will the results be depending on which molecule you choose? Do they all change at the same rate (over evolutionary time)?
- Most of the databases are very "messy" and hard to sort through. Thus it is hard to be sure you have found exactly what you are looking for. Ask your advisor to check with you! The same is true for many of the online analysis tools.
- Since most of your "experiments" are done on the computer with computational tools found at biology related websites, it is VERY important that you understand (at least in very general terms) WHAT the programs do and what you learn from using them.
Terms and Concepts
- DNA, gene
- mRNA
- Amino acid, protein sequence
- Protein, translation
- Coding sequence
- Protein domain
- Protein motif
- Sequence/protein/molecular evolution
- Homolog/homology
- Sequence alignment, distance matrix
- Eukaryote, prokaryote
- Phylogenetic/evolutionary tree
Bibliography
Background knowledge/info- NHGRI Education: About Genomics. Retrieved September 10, 2021.
- Cold Spring Harbor. (n.d). DNA Learning Center. Retrieved September 10, 2021.
- ENSI. (n.d.). Evolution Lesson. Indiana University.Retrieved September 10, 2021.
- PBS Tutorial: Evolution (particularly the Online Lessons for Students) Retrieved September 10, 2021.
- Access Excellence. (n.d.). Activities Exchange - Evolution. The National Health Museum. Retrieved September 2, 2021.
- Popular science/media stories
- NCBI. (n.d.). OMIM. Online Mendelian Inheritance in Man. Retrieved September 2, 2021.
- NIH. (n.d.). PubMed (searchable database of scientific journal articles). National Library of Medicine. Retrieved September 2, 2021.
- NCBI. (n.d.). Welcome to NCBI. National Center for Biotechnology Information.
Retrieved September 2, 2021.
- Can access OMIM, BLAST, GenBank, PubMed from here and search databases of protein and DNA sequences.
- NCBI. (n.d.). BLAST. National Center for Biotechnology Information. Retrieved September 2, 2021.
- NCBI. (n.d.). COG. National Center for Biotechnology Information. Retrieved September 2, 2021.
- EMBL-EBI. (n.d.). T-Coffee. Retrieved September 2, 2021.
- Has useful colored output (click "html" under Scores column of results page). Note that sequences must be in FASTA format (see help file).
- EMBL-EBI. (n.d.). ClustalW2. Retrieved September 2, 2021.
- Don't worry about all the optional parameters -- simply paste in all of your sequences in FASTA format.
- Prosite (database of protein families and domains): Prosite. Retrieved September 2, 2021.
- CDD (collection of protein domains derived from several sources): Conserved Domains. Retrieved September 2, 2021.
- NCBI. (n.d.). Structure: Cn3D installation. Retrieved September 2, 2021.
- Power + Bulk. (n.d.). Protein Explorer. Visualization of protein structures. Retrieved September 2, 2021.
Experimental Procedure
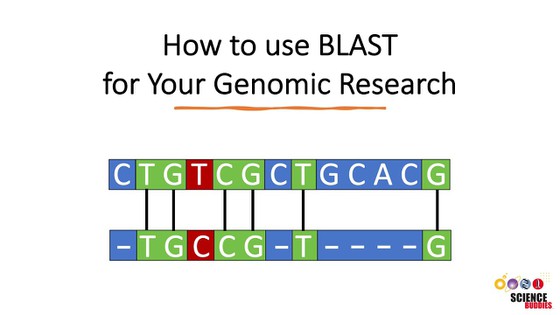
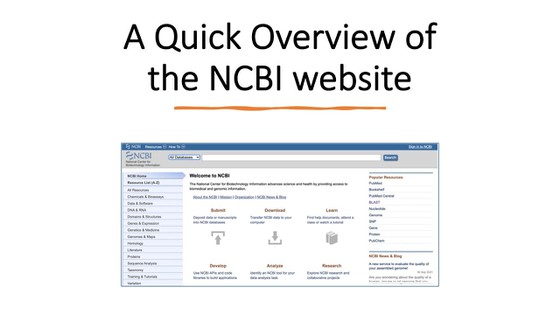
- Before you start with this project, it might be helpful to familiarize yourself with the bioinformatic tools and websites that you are going to use. You can watch the two videos below to learn more about the BLAST tool and the NCBI website and databases.
- Identify a few genes/proteins of interest. You may choose a gene/protein you heard about through a major media source like the news or a magazine. One other good resource is a database called "Online Mendelian Inheritance in Man" (OMIM) which catalogs all known human genetic diseases and which genes may be involved in causing or creating susceptibility to that disease (if that information is known). You can also try to use "PubMed" which is a searchable database of scientific journal articles on as many topics as you can think of, but these articles are usually pretty dense and tough to read—but you can learn a lot from reading the abstracts available for every paper. Remember to choose a few as not all proteins are found in all species, nor is sequence data available for many of the species that have that protein. Also, think about which "type" of protein you are choosing—you will want at least one of the genes that you study to exist in distantly related organisms.
- Get the amino acid (protein) sequence and the mRNA sequence for your protein of interest. To get the amino acid sequence, go to the NCBI website and search the "protein" category for the name of the gene/protein that you have chosen. Notice that sometimes your gene may be listed under an abbreviation or slightly different spelling—so just keep trying variations until you get it. To get the mRNA sequence, repeat your search using the "nucleotide" category. This search gives you both DNA and mRNA sequences. You can either sift through them manually to find mRNA sequences, or click on "mRNA" in the "Molecule types" section on the left side of the page to get only mRNA sequences. Save the amino acid and mRNA sequences for your gene in FASTA format (described below). Get help from your advisor making sure you've got the "whole protein" as you will probably get more than one "hit" when you do your search. Be sure to read the sections on "Tips for Searching Genbank" and "Tips for Formatting Sequences" below to guide you in your data collection. And, be sure to document the accession number of each protein you select. (See definition of accession number below.)
- Use the BLAST program (at NCBI or elsewhere) to search the non-redundant (nr) database of proteins (=BLAST-P) for homologs from other species. All BLAST does is search all available databases for other proteins that are the most similar to the one you entered. It then gives you an output with "matching" sequences ranked by which is the most similar to the one you entered. Be sure to check the alignment on the BLAST readout to make sure the two sequences don't just share a small stretch of similarity, but look to actually be from homologous proteins. You can check that in the "Graphic Summary" section of the BLAST result page. Try to find your gene in at least 3 or 4 other organisms in order to construct a meaningful sequence alignment. (Note: Some programs "max out" if you try to put too many sequences in, so you may have to do them in batches.)
- Use a multiple sequence alignment program (see tools above) to do a "multiple sequence alignment" to compare sequences from multiple species. Do this separately for mRNA and protein sequences, and compare your results. Do you expect one to show more differences than the other? Your input file should be a list of FASTA formatted sequences representing the same gene in different organisms. All this kind of program does is line up the proteins (using their most similar regions) so that you can see how similar the sequences are to each other at each position. The output will show you how conserved each part of the sequences are across the species that you used—some even color code the degree of similarity. Based on your alignment, which parts of the protein do you think are important. How might you test your hypothesis?
- Explore the function(s) of your proteins using a domain identification database (see online tools listed in bibliography). These tools typically require you to input your amino acid sequence in FASTA format. Learn a little about some of the domains that you identify—particularly any that occur in the regions that you have shown to be conserved. Can you see how do the functions of these domains relate to the overall job of the protein? How do your results here relate to the results of your sequence alignment?
- For making a better query, try asking for both the name of the protein and the organism from which you would like that sequence. (For example, try entering "myoglobin homo sapiens" to look for the human version of the myoglobin protein sequence.)
- When sorting through the results, many entry titles are followed by two Latin words in parentheses such as (Homo sapiens). These words represent the scientific name of the animal from which the sequence came.
- Some animals are used very often in genetic research and are called "model organisms." You are very likely to run across sequences from these organisms. See the table of some of the commonly used model organisms and their scientific names.
| Common Name | Scientific Name |
|---|---|
| Human | Homo sapiens |
| Mouse | Mus musculus |
| Rat | Rattus norvegicus |
| Zebra Fish | Danio rerio |
| Fruit Fly | Drosophila melanogaster |
| Round Worm | Caenorhabditis elegans |
| Yeast | Saccharomyces cerevisiae |
- The genomes of many other animals are being sequenced (mainly) for sequence comparison studies such as the one you are performing. Here are a few of these types of organisms you might run across. If the name you find is not on this list, just do a web search and see if you can figure out to which animal the name refers.
| Common Name | Scientific Name |
|---|---|
| Chimpanzee | Pan troglodytes |
| Baboon | Papio anubis |
| Rhesus Monkey | Macaca mulatta |
| Cow | Bos taurus |
| Dog | Canis familiaris |
| Cat | Felis catus |
| Opossum (laboratory) | Monodelphis domestica |
| Pig | Sus scrofa |
| Chicken | Gallus gallus |
| Frogs | Xenopus laevis and Xenopus (Silurana) tropicalis |
| Puffer Fish | Takifugu rubripes |
| Purple Sea Urchin | Strongylocentrotus purpuratus |
| Acorn Worm | Saccoglossus kowalevskii |
| Honey Bee | Apis mellifera |
- These queries are very literal and just look for your search words anywhere in the title or description of the sequence. So if you enter "myoglobin," you may get a hit to a protein described as a "myoglobin binding protein" or "similar to myoglobin," neither of which is what you are looking for. You can get around this by using the "Advanced" search option: Click on the word "Advanced" below the text box where you enter your gene or protein name. In the new window, go to the pull down menu that says "All Fields" and change it to "Title." This way, your search term has to appear in the title of the database entry. You still may get multiple hits, so just keep scrolling through the results until you find the myoglobin protein itself.
- Some sequence entries represent fragments or pieces of a protein that have been important for one experiment or another. For this project, you need the whole sequence, so try to avoid using sequences that have the words partial, chain or subunit in them. If all of the entries seem to be incomplete, you may need to choose a different protein.
Note: If you are doing a project that involves searching for DNA/mRNA sequences, some entries will be marked as either "partial cds" or "complete cds," where cds stands for "coding sequence." You should choose "complete cds" if such an entry can be found for your gene—remember this point applies only to DNA/mRNA searches. - Finally, what is the best way to refer to the "right" sequence when you find it? The first string of letters/numbers in the output of any search on NCBI is called the accession number. This may look something like NP_00555 or AAH14547. This is just a unique identifier for that sequence file in that database. So you could tell anyone else in the world you are using the sequence from accession number NP_00555 and they would be able to access the exact same record. If you want to get back to that file, you can just enter that accession number for your query and the proper file should come up every time.
- One other thing you might run in to is cDNA. What is a cDNA? A cDNA is just the sequence of a DNA string that would be COMPLEMENTARY (thus the "c") to an mRNA of interest. (RNA molecules are inherently unstable, so scientists often make a DNA copy of an RNA to make experiments easier.) So a cDNA sequence represents an mRNA found in a cell.
- Are you looking at DNA or protein sequence? If the string of letters in your sequence is made up of a, t, g and c, then you are looking at a DNA or RNA sequence. If the string looks like "random" letters from the alphabet (like MQPLLG), then you are looking at a protein sequence. Each amino acid has been assigned a single letter code to represent it. (You can find this code online or in a textbook.)
- When copying your sequence of interest to save it or enter it into another program, there are a couple of ways you can do it. Whichever way you choose, it is probably easiest to "store" and manipulate the sequence in a word processing program. You can then save it as a "text" file so that it doesn't have any weird word processing formatting. (You may also be able to upload a text file into a DNA analysis program.)
- The first way to save the sequence is just using the string of sequence alone. From the accession number file, you can just copy and paste the letters onto a word processing document. Then just take out any of the numbers that were copied over. You don't need to worry about spaces and returns because the programs you will be using are trained to ignore them and only read the "letters" in the file. Then, you can just copy and paste this saved text into the program you are using.
- Most programs that analyze DNA/mRNA/protein sequences require them to be in a certain format. Thus, when you are saving the sequences that you plan to use in your later analysis, you should save them in the conventional format. The most common format is called "FASTA" format. FASTA files are structured with the ">" symbol followed by the name of the sequence (like myoglobin), then a return and the text of the sequence. Most of the programs you use will recognize a FASTA file and know that what comes after the > and before the first return is the "name" of the sequence. Here is an example of a FASTA format sequence:
>Myoglobin protein mglsdgewql vlnvwgkvea dipghgqevl irlfkghpet lekfdkfkhl ksedemkase dlkkhgatvl talggilkkk ghheaeikpl aqshatkhki pvkylefise ciiqvlqskh pgdfgadaqg amnkalelfr kdmasnykel gfqg
- The simplest way to get your sequence into FASTA format is to use the FASTA format display option in Genbank. Once you're on the page containing the DNA or protein sequence that you want, you can get the sequence into this format by clicking on the "FASTA" link underneath the GenBank number at the top of the page (below the sequence title). You can then either cut and paste the formatted sequence into a text file, or use the pull down menu next to the "Send To" button and select "File." If you then click on the "Create File" button, it will download the sequence on your screen to the file name you specify.
Note that you don't need to worry about spaces and returns in most sequence analysis programs because they ignore them and only read the "letters" in the file.
Ask an Expert
Global Connections
The United Nations Sustainable Development Goals (UNSDGs) are a blueprint to achieve a better and more sustainable future for all.

Variations
- Run the protein through a 3D structure prediction program.
- Search for literature on effects of mutations in different parts of the protein. OMIM is a good source for mutations if you have chosen a protein involved in a human disease. PubMed is a good source for the results of experiments of scientists studying particular proteins—perhaps there are papers where someone has tried mutating your protein of interest. How could this help you?
- Try translating your human mRNA sequence into the correct protein sequence using an online translator (try http://us.expasy.org/tools/dna.html). Does your output match the human amino acid sequence that you obtained from Genbank? If not, why might this be the case? Can you identify the "start codon" (the place where translation begins) and the "stop codon" the place where translation ends? What's left on the mRNA on either side is called 5' and 3' untranslated region (UTR), which can be important in regulation, transport, and processing of the mRNA.
- Try retrieving the DNA sequences for one or more of you proteins of interest from humans and several non-humans. How does your DNA alignment look different from your protein and mRNA alignment? Why might this be the case? Hint—think about introns and exons!
Careers
If you like this project, you might enjoy exploring these related careers: